Faced with the likelihood bout of irregular heartbeats might finish his life, in 2006, Dr. Marc Sicklick had a small gadget implanted in his chest that will shock it again into rhythm. Soon he would battle with one other life-or-death alternative: whether or not to take away the Sprint Fidelis, which was deemed harmful and recalled in 2007 after it had been implanted in a whole lot of 1000’s of sufferers.
The Sprint Fidelis was susceptible to giving sufferers random electrical jolts — and typically failed to fireplace in real cardiac emergencies, in response to producer Medtronic’s letter to doctors.
What Sicklick and 1000’s of others in his place haven’t identified is that the Food and Drug Administration quietly took steps to maintain crucial info out of the general public gentle. Shortly after the recall, the FDA and Medtronic made a deal to maintain studies in regards to the broadly used gadget’s malfunction incidents — now totaling 50,000 — shielded from public scrutiny.
The FDA has allowed gadget makers to file 1.1 million studies of accidents or malfunctions to a little-known inner FDA database since 2016, a current Kaiser Health News investigation has found, spurring high FDA officers to pledge to open these data inside weeks and shut down the “alternative summary reporting” program.
For the previous twenty years, the company has granted varied units various kinds of so-called exemptions from reporting to their public-facing database, known as MAUDE, KHN has discovered.
One of these exemptions coated the Sprint Fidelis. In May 2008, it was granted a “remedial action” exemption — allowed when “the manufacturer has initiated reasonable and appropriate actions to mitigate the problem(s)” and additional studies of hurt is not going to “provide any significant new data,” FDA spokeswoman Deborah Kotz stated in an e mail. She stated the FDA hasn’t granted such an exemption since 2015 and has “effectively ended the program,” and couldn’t say whether or not that knowledge could be opened to the general public quickly.
The logic of the FDA’s method sits poorly with sufferers like Sicklick who’ve a Sprint Fidelis buried deep of their chests. “Worrying about it not working [has] caused a tremendous, tremendous amount of anxiety in me, and I’m not an anxious person,” stated Sicklick, an allergy specialist from Long Island, N.Y. “I think the FDA should be making this information public.”
Six high cardiologists interviewed for this report stated they weren’t conscious the FDA had granted Medtronic such an exemption.
“Amazing. Really amazing,” stated Dr. Robert Hauser, the Minnesota heart specialist whose analysis first introduced the excessive fee of Sprint Fidelis failures to gentle. “It’s not in the best interest of the patients who have these devices.”
Hauser and different medical doctors have spent years serving to sufferers navigate the dangers of dwelling with a cardiac implant that might hurt them or present process a sophisticated device-removal surgical procedure that might show lethal.
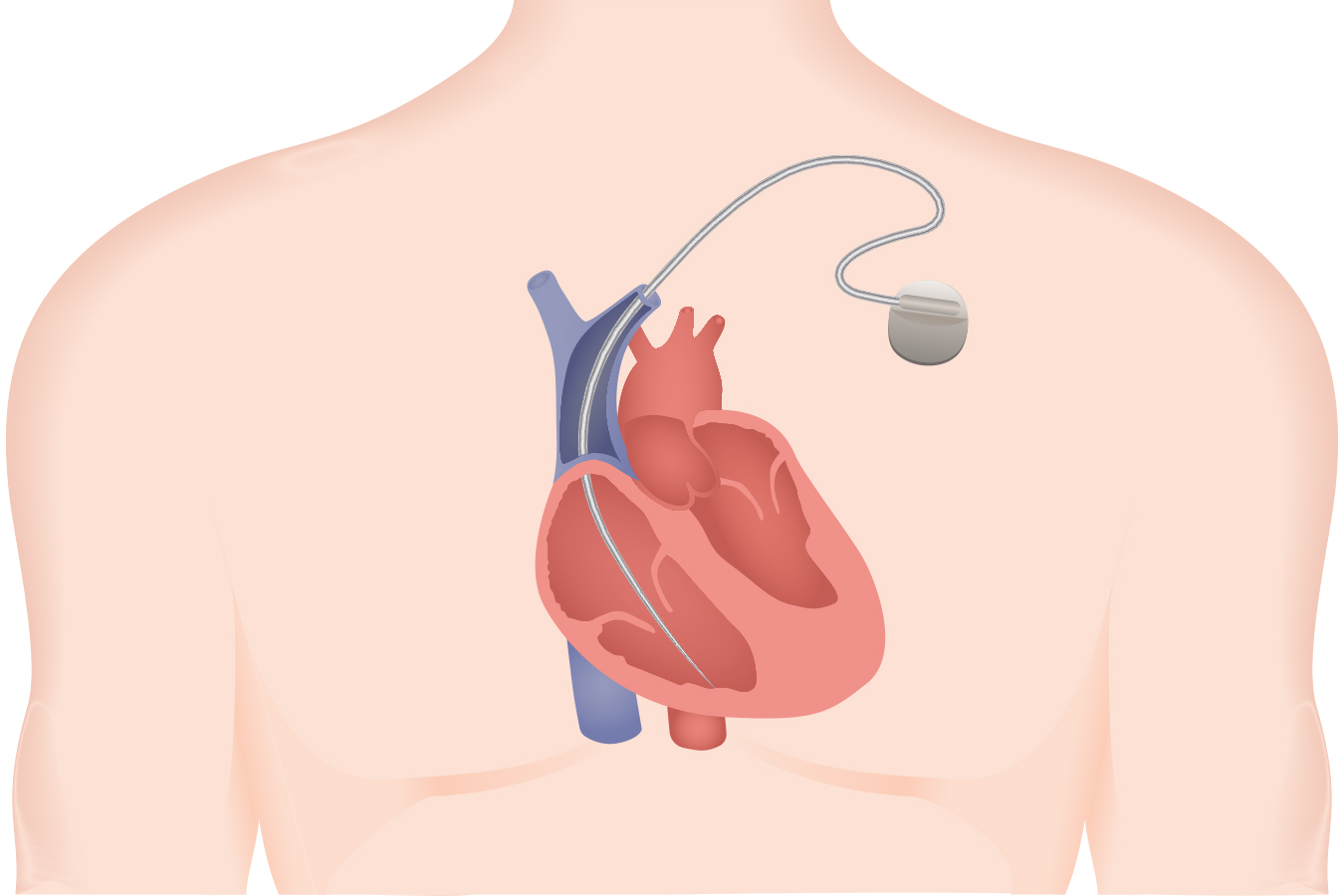
The gadget is a pair of wires tied to a defibrillator designed to shock the center again into a traditional rhythm. By the time it was recalled in late 2007, the Sprint Fidelis had been implanted within the coronary heart tissue of 268,000 sufferers.
While medical doctors and sufferers can voluntarily report issues with the gadget, the FDA requires gadget makers to report back to MAUDE the accidents they change into conscious of — except they’ve an exemption.
Through March, Medtronic says, it logged greater than 50,000 cases of hurt or malfunction, with a couple of downside reported for among the 35,000 instances.
While just a few reports within the MAUDE database seek advice from the existence of an exemption for the Sprint Fidelis, the FDA has stated these “remedial action” studies are filed internally and obtainable to the general public via a Freedom of Information Act request, which may take almost two years to course of.
Medtronic, which equipped knowledge on the exemption for this story, stated that its Independent Physician Quality Panel has been reviewing knowledge on the gadget since 2007 and that the knowledge has been periodically reported to physicians. “Patient safety is our top priority,” spokesman Jeffrey Trauring stated in an e mail.
Trauring stated his firm operates the “most comprehensive [defibrillator] lead surveillance program in the industry.” He additionally stated Medtronic has updated information for doctors in regards to the lead’s efficiency with the “most reliable data,” as in contrast with the FDA’s open MAUDE database and its “significant and well-established limitations.”
Kotz stated by way of e mail that the FDA has “worked closely with Medtronic” to observe the efficiency of the recalled leads and “ensure appropriate patient management recommendations are available to clinicians” in twice-yearly performance reports.
“[Medtronic provides] additional information on product performance and safety in a more comprehensive format than what we receive in summary reports,” Kotz stated.
None of the Medtronic publications, although, provide particulars in regards to the 50,000 malfunction and hurt studies the corporate lodged with the FDA over 10 years. Those until-now-unacknowledged studies outline the variety of lead fractures or cases of inappropriate shocks.
The Medtronic studies tally much less particular “advisory related events.” They additionally monitor extra detailed malfunctions for a smaller variety of units both shipped again to the corporate or enrolled in a examine.
Detective Work
A dozen years in the past, earlier than Sprint Fidelis was recalled, Hauser was a working towards heart specialist on the Minneapolis Heart Institute when two sufferers in a single weekend arrived with fractured Sprint Fidelis leads. Hauser stated he appeared on the middle’s information and located 5 comparable instances.
Then he delved into MAUDE and located much more instances. He known as Medtronic. He printed a paper about what he had discovered, which he stated “went viral.”
On Oct. 15, 2007, Medtronic issued a recall, saying the gadget gave some sufferers inappropriate shocks and was a “possible or likely” contributing consider 5 affected person deaths. The firm later revised the demise rely to 13.
The thinnest defibrillator lead marketed to this point, researchers stated the Sprint Fidelis wires had been susceptible to ship defective alerts to the defibrillator, inflicting affected person accidents.
In its 2007 letter to doctors, Medtronic suggested in opposition to surgically eradicating the lead, citing charges of main problems, together with demise, starting from 1% to 7%.
The recall was large information. Patients would testify to Congress or inform journalists about surprising shocks that felt like a cannon to the chest or being kicked by a horse.
Then-House oversight committee chairman Henry Waxman fired off a letter to the FDA per week after the recall, partly asking in regards to the firm’s reporting of antagonistic occasions.
The company acknowledged in a Dec. 13 response letter that Medtronic was reporting “the majority of adverse events” on their implantable defibrillators to the FDA “in aggregate electronically, via compact discs.”
Medtronic’s Trauring stated on the time the corporate had an “alternative summary reporting” exemption for instances of lead infections or erosion, which meant the wires had moved out of place contained in the physique.
Six months later, on May eight, 2008, the FDA gave Medtronic a second exemption, permitting the corporate to report issues central to the recall outdoors of public scrutiny.
Trauring stated in an e mail that underneath that second “remedial action” reporting exemption, Medtronic reported a complete of 36,914 instances with 50,205 problems.
They embrace greater than 22,000 cases of lead fracture and almost 2,900 incidents of inappropriate shocks. Trauring stated the corporate discontinued the abstract reporting on Nov. 1, 2018, attributable to low volumes of points.
Death instances weren’t filed with the studies granted exemptions. Since the 2007 recall, greater than 2,300 studies of deaths during which the gadget was suspected to have performed a job had been filed with the FDA and are publicly obtainable, data filed in MAUDE present. A 2009 report says one affected person had 13 painful shocks after which bleeding throughout a surgical procedure to take away his defective lead. An legal professional reported to the FDA that inside months that affected person died.
In 2012, a affected person with a defective gadget died after a serious vessel was torn throughout a surgical procedure to take away the lead. In 2015, a affected person went to the emergency room after getting 5 shocks, was deemed to be in coronary heart failure “exacerbation” and died within the hospital 4 days later, studies in MAUDE present.
Meanwhile, researchers making an attempt to measure the hurt ensuing from the defective gadget had been reaching totally different conclusions. While Medtronic-backed research had been reporting a three-year lead survival fee of 95% to 97%, Hauser and his colleagues discovered a decrease fee, 88%, in response to a 2010 article within the Netherlands Heart Journal.
Dr. John Mandrola, a heart specialist at Baptist Medical Associates in Louisville, Ky., stated the Sprint Fidelis was the “worst cardiac device problem” he has seen in 22 years of apply. He stated a few of his sufferers had been traumatized by surprising shocks and the repercussions went on for years. That the FDA didn’t publicly report ongoing hurt “seems problematic to me,” he stated. “What is the benefit to the public of an exemption?”
Dr. Frederic Resnic, cardiovascular medication division chairman at Lahey Hospital & Medical Center in Burlington, Mass., and a Tufts University medical college professor, has testified to Congress about gadget security. He stated in an e mail that he commends the FDA for current work to modernize security surveillance for medical units, however the “lack of communication and transparency” over the Sprint Fidelis exemption “challenges the FDA’s unique role as the primary, trusted, information source regarding medical device safety.”
Dr. Rita Redberg, a University of California-San Francisco cardiology professor and editor of JAMA Internal Medicine, stated the FDA has acknowledged that device-related issues are woefully under-reported.
“Now the FDA is making it worse intentionally,” she stated. “I can’t think of a justification for it, and I think it’s a dangerous practice.”
KHN knowledge correspondent Sydney Lupkin contributed to this report.